
What we do
See blog posts on Andrew’s personal website.
Explanation of research for possible student positions can be found in student_projects.
Code snippets and recipes are also available on GitHub Gists.
Low-cost computational tools and robust software for decision-making
We are working toward bridging the gap between how computational tools are used in chemical and materials design processes. A significant part of this is the development of new software ecosystems that are FAIR and easy-to-use by researchers of various expertise.
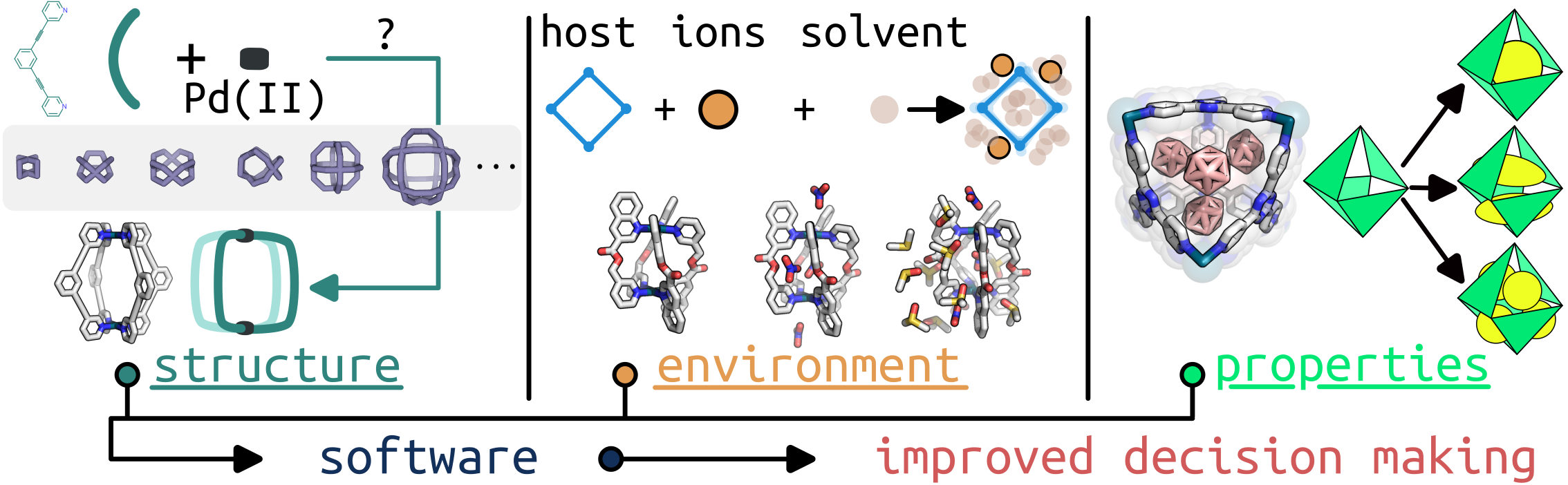
Structure generation of (supra)molecular materials
We continue to develop an ecosystem built around the work done by Andrew in stk for generating molecular and periodic structures. Find tutorials: stk tutorial videos and code.
Using coarse-grained and minimal models
cgx is a library for using stk with coarse-grained molecules to afford more efficient high-throughput screening and rational design. This library is under-development but it’s first usage is written up here.
We recently introduced the use of minimal model and atomistic model structure prediction with cgx in two works: firstly, predicting starhsips and then a series of case studies.
Preparing building blocks for structure generation
bbprepared contains algorithms and tests for preparing building blocks for stk usage. Here is a video on its usage. The documentation contains useful recipes and examples, such as finding the lowest energy conformer:
import stk
import stko
import bbprep
building_block = stk.BuildingBlock(smiles="C1=CC=NC(=C1)C=NBr")
# This uses the rdkit conformer generation.
generator = bbprep.generators.ETKDG(num_confs=100)
ensemble = generator.generate_conformers(building_block)
# Iterate over ensemble.
minimum_score = 1e24
minimum_conformer = bbprep.Conformer(
molecule=ensemble.get_base_molecule().clone(),
conformer_id=-1,
source=None,
permutation=None,
)
for conformer in ensemble.yield_conformers():
# Something here to calculate energy.
score = stko.MMFFEnergy().get_energy(conformer.molecule)
if score < minimum_score:
minimum_score = score
minimum_conformer = bbprep.Conformer(
molecule=conformer.molecule.clone(),
conformer_id=conformer.conformer_id,
source=conformer.source,
permutation=conformer.permutation,
)
Optimising structures
stko contains wrappers for optimisation and property calcualtion of stk molecules. The documentation contains recipes and tutorials.
I have also developed some low-cost Monte Carlo-based algorithms for optimisation stk molecule optimisation (MCHammer) and host-guest conformer generation/optimisation (SpinDry).
Visualising molecular pores
PoreMapper is a very efficient way to map the inside of a molecular pore for visualisation and analysis. See a blog post here.
Databases of toy-model material space
While the cgx documentation holds examples and tutorials for usage, we also publish our toy-model landscapes on cgmodels, which are explorable using chemiscope.
Exploring host-guest system design at low-cost
Calculating molecular size
mol-ellipsize is an efficient method for calculating molecular size based on fitting an ellipsoid to the vdw cloud of a molecule. Examples are provided within the github repository. See a blog post here.
Interpreting and analysing dynamic trajectories
dynsight is a software package Andrew manages the development of in collaboration with the Pavan Lab. This software is working toward streamlining the analysis of trajectories of particles. A recent write up can be found here